气相色谱一质谱法测定枸杞中28种农药残留
 材料与方法
材料与方法
1.1材料与试剂
earb/NH2固相萃取柱(500 mg/500 mg/6 mL,德国CNW公司);丙酮、正己烷(色谱级,美国J.T.Baker公司);乙腈(色谱级,德国Merck公司);甲苯(色谱级,德国CNW公司);敌敌畏、敌百虫、乙酰甲胺磷、甲拌磷、氧乐果、久效磷、乐果、百菌清、毒死蜱、对硫磷、三唑酮、马拉硫磷、三氯杀螨醇、丙溴磷、联苯菊酯、甲氰菊酯、氯氟氰菊酯、氟氯氰菊酯、哒螨灵、氯氰菊酯、氰戊菊酯、溴氰菊酯、苯醚甲环唑、二甲戊乐灵、克螨特、丙环唑、戊唑醇、咪鲜胺均购自中国国家标准物质中心,浓度均为1 000 mg/L。其余试剂为分析纯,试验用水为超纯水。
1.2仪器与设备
Agilent7890A气相色谱仪,7000B Triple Quad GC/MS质谱仪(Agilent公司);MassHunter数据处理软件;Rapid Trace全自动固相萃取工作站(美国Biotage公司);MS3 basic涡旋混合器(德国IKA公司);T18 ULTRA TURRAX(德国IKA公司);R-210旋转蒸发器(瑞士步琦公司);KQ-500DB超声波清洗器(昆山市超声仪器公司)。
1.3方法
1.3.1溶液配制 单标储备液的制备:分别量取标准溶液各1.0 mL,用丙酮稀释成100 mg/L的标准储备液,-20℃下保存备用。混合标准工作液的制备:吸取一定量的各标准储备液置于同一容量瓶中,用丙酮稀释成合适浓度的工作溶液。
1.3.2样品前处理 称取(5.00±0.01)g枸杞于50 mL离心管中,加入5 mL水和15 mL乙腈,15 000 r/min匀浆提取1 min,加入2 g氯化钠,涡旋混合1 min,6 000 r/min离心5 min,取上层有机相至浓缩瓶中。离心管中再加15 mL乙腈,重复提取1次,抽取并合并上层有机相,40℃旋转蒸发至1~2 mL,待净化。earb/NH2固相萃取柱用4 mL乙腈+甲苯(体积比3:1)预淋洗,待液面到达柱吸附层时,迅速将上述待净化液转移至净化柱上,再用2 mL乙腈+甲苯(体积比3:1)洗涤样液瓶并转移上柱,重复3次,用25 mL乙腈+甲苯(体积比3:1)洗涤净化柱,收集流出液于浓缩瓶中,40℃旋转蒸发至近干,加入5 mL正己烷再旋转蒸发至干,用正己烷+丙酮(体积比1:1)定容至1 mL,过0.22μm有机相滤膜,用于气质测定。
1.3.3色谱、质谱条件 色谱条件:色谱柱:DB-17MS(30 m×0.25 mm×0.25 μm,美国Agilent公司);升温程序:初始温度70℃,恒温1 min,以8℃/min的速率升温至210℃,再以20℃/min的速率升温至290℃,保持12 min;进样口温度280℃;进样方式:脉冲不分流;载气:高纯度氦气;流速:1.2 mL/min;进样量:1.0 μL。質谱条件:电子轰击(EI)电离模式;电离能量70 eV;扫描方式:选择离子监测(sIM);离子源温度:230℃;四级杆温度:150℃;GC-MS接口温度:280℃;溶剂延迟:3.75 min。
1.3.4标准曲线、方法检出限及定量限 将28种农药混合溶液稀释成10.0、5.0、2.0、0.5、0.2、0.1、0.05、0.02、0.01、0.002μg/mL系列标准工作液,采用“1.3.3”节的方法进行分析,以进样质量浓度为横坐标,峰面积为纵坐标,进行线性回归计算,求得28种农药的线性方程、相关系数。在阴性样品中添加28种农药标准品,计算3倍信噪比(S/N>~3)和10倍信噪比(S/N≥10)时所对应的样品质量浓度,分别作为方法的检出限(LOD)、定量限(LOQ)。
1.3.5方法回收率及精密度试验 采用对阴性样品进行加标回收试验来考察方法的准确度、精密度,对空白枸杞样品,分别添加不同浓度的农药混合标准溶液,按照“1.3.2”节的方法进行样品前处理,在“1.3.3”节条件下测定,计算样品加标回收率。每个添加水平重复6次,测得各组分的峰面积,计算其相对标准偏差。
2结果与分析
2.1色谱条件优化
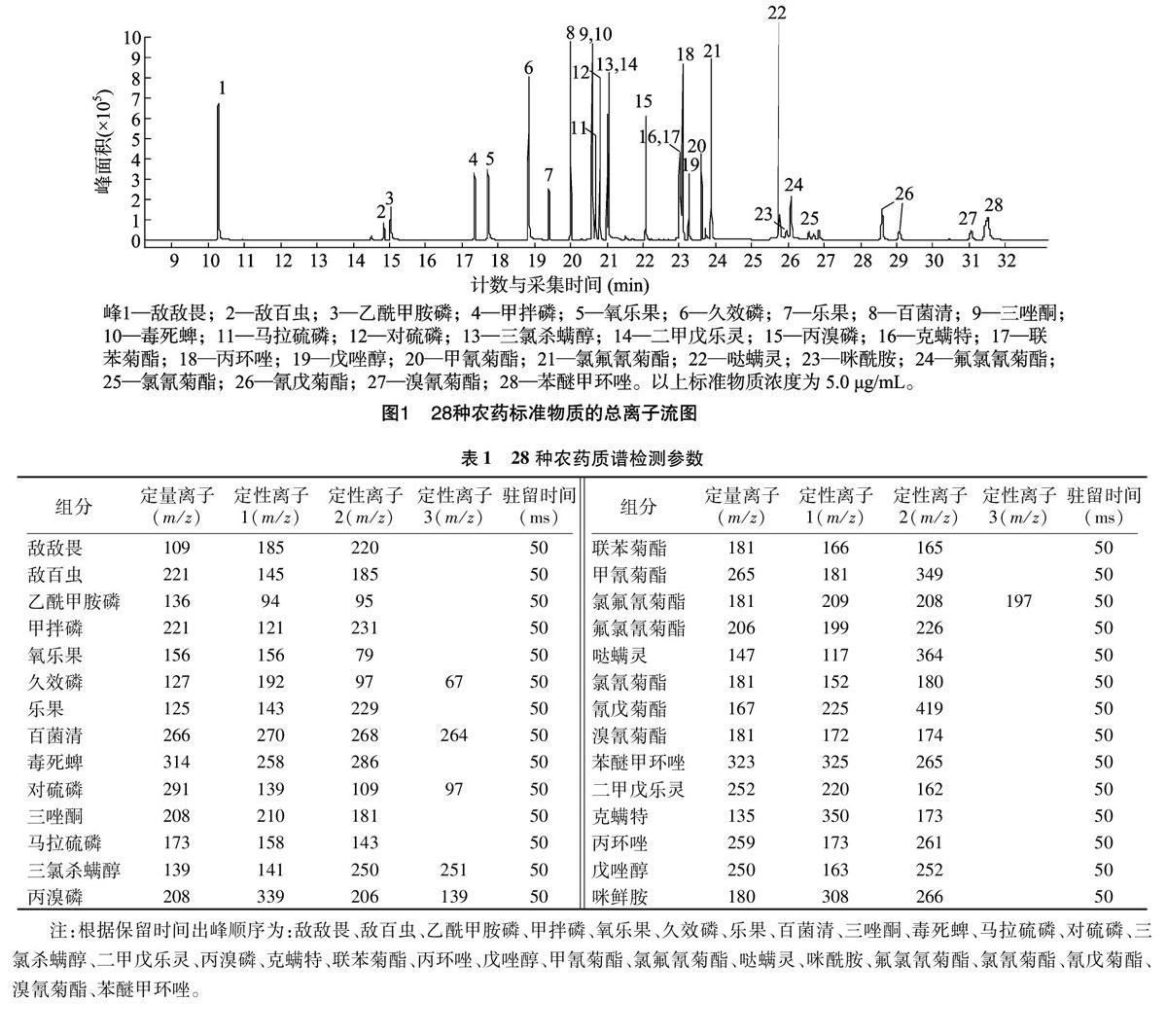
选择合适的色谱柱是农药多残留分析的前提,本试验考察了DB-5MS、DB-17MS等2种不同极性的色谱柱,通过标准工作液进样来比较两者对28种农药的分离效果。结果显示,DB-5MS、DB-17MS均能实现组分的基本分离,但乙酰甲胺磷、久效磷等有机磷农药在DB-17MS色谱柱上的峰形(图1)优于DB-5MS色谱柱。
22
2.2质谱条件优化
按照出峰顺序对测物进行分组,分时段完成所有离子的检测。保留时间接近的化合物被分进一组进行多离子同时扫描。采用SIM方法,每种化合物分别选择1个定量离子、2~3個定性离子,以响应值较高且干扰较少的离子进行定量,以特征离子及离子问的相对丰度比进行定性。28种化合物优化的定量离子及定性离子等参数见表1。SIM扫描每个组分定量离子流图见图2。
2.3样品前处理条件优化
比较了乙腈、丙酮、丙酮+正己烷(5:5)等溶剂对28种农药的提取效果,结果表明,枸杞复水后加乙腈提取效果最好,这可能与样品中加入水促进了枸杞中糖分溶解,从而提高了乙腈的渗透能力有关。加入NaCl后,乙腈可从混合溶液中快速分离,有利于后续操作。丙酮提取液中干扰物较多,不利于下一步净化。丙酮+正己烷(5:5)的提取效率低于以上2种提取溶剂。因此,本试验采用乙腈作为提取溶剂。
目前多农残分析主要采用固相萃取柱吸附杂质方式来净化,本试验对比了C18、Florisil、carb/NH23种吸附剂的净化效果。由于枸杞基质复杂,含有较多的糖类、蛋白质及色素等,C18对这些杂质的吸附能力弱,净化效果较差。Florisil柱虽然对杂质有较强的吸附能力,但对极性较强的有机磷农药也有吸附能力,增强洗脱液极性,可将此类组分洗脱,但杂质也会被同步洗脱。carb/NH2的净化效果最好,但对百菌清等含有芳香族结构的农药也产生明显吸附效果,在洗脱液中加入25%左右的甲苯可以破坏吸附剂和此类农药的相互作用。同时,甲苯的存在可防止碱性敏感的农药在碱性吸附剂(NH2)上的降解。因此,本试验选择carb/NH2作为固相萃取柱,乙腈+甲苯(体积比3:1)为洗脱溶剂。
2.4标准曲线、方法检出限和定量限
28种农药混合标准溶液,采用外标法定量,在“1.3.3”节条件下分析,以定量离子峰面积(y)对质量浓度(x)得出校正曲线,通过回归分析得出回归方程、决定系数(表2)。结果表明,28种农药在0.01~5.00μg/mL范围内线性关系良好(r2>0.99),方法的检出限(LOD)和定量下限(LOQ)分别为0.008~0.085μg/g和0.027~0.280μg/g,满足农药残留定量分析要求。
2.5方法的回收率与精密度
分别在空白样品中添加不同浓度水平的混合农药标准工作溶液,每个添加水平平行分析6次,添加后按“1.3.2”节和“1.3.3”节所述步骤进行提取、净化、测定,样品加标回收率、相对标准偏差见表3。结果显示,在3个加标水平下,28种农药的平均回收率范围为71.0%~106.0%,相对标准偏差为0.96%~12.3%,表明该方法准确可靠。
2.6实际样品测定
利用本试验方法,分别对来自农贸市场、超市、农户、枸杞加工企业、生产基地的27个枸杞样品进行检测,其中有11个样品检出23次农药残留。检出的农药和检出量分别为:毒死蜱(0.091~0.275 mg/kg)、丙溴磷(0.528 mg/kg)、联苯菊酯(0.016 mg/kg)、三氟氯氰菊酯(2.22 mg/kg)、哒螨灵(1.09 mg/kg)、氯氰菊酯(0.194~0.434 mg/kg)、氰戊菊酯(0.284~0.616 mg/kg)、溴氰菊酯(3.11~3.59 mg/kg)、苯醚甲环唑(0.0780~0.212 mg/kg)、甲氰菊酯(0.148 mg/kg)、克螨特(0.145 mg/kg)、戊唑醇(0.412 mg/kg)。
3结论
本研究建立了枸杞中28种农药多残留的气相色谱一质谱联用检测方法,包括11种有机磷杀虫剂、1种有机硫杀虫剂、1种有机氮杀虫剂、3种有机氯杀虫剂、7种拟除虫菊酯类杀虫剂、4种含氮杀菌剂、1种除草剂,该方法灵敏度高、结果准确可靠,重复性好,为枸杞中农药多残留的测定提供了简单可靠的前处理方法和检测手段。
上一篇:粮食短缺的救星:有机农业
下一篇:锐胜处理姜种效果试验初报